

【本月之星】AMPK、TREM2、p53 基因敲除细胞
想设计别树一帜的研究实验计划获取高额基金支持吗?想知道最新的研究热点保持自己的学术敏锐度吗?想又快又便宜地获取实验所需细胞吗?这里,小编根据本月的市场热点,整理了本月的现货之星,希望对大家的科研之路有所启发
一、AMPK基因敲除细胞
该基因编码的蛋白质属于 ser/thr 蛋白激酶家族。它是 5'-prime-AMP 活化蛋白激酶 (AMPK) 的催化亚基。AMPK是一种在所有真核细胞中保守的细胞能量传感器。AMPK 的激酶活性被细胞 AMP/ATP增加的比率刺激激活。AMPK通过磷酸化调节许多关键代谢酶的活性。它通过关闭消耗ATP的生物合成途径来保护细胞免受导致ATP消耗的压力。已经观察到编码不同亚型的可变剪接转录物变体。
肾上腺素能系统是增强心藏血液输出量的有效刺激,当能量代谢受损时,可能会变得有害。今年9月法国巴黎萨克莱大学实验团队在PloS one期刊上发表的文章研究了在能量消耗时被激活的AMP激活蛋白激酶(AMPK)是否可能控制-肾上腺素能途径。本文研究了AMPKα2基因敲除小鼠对肾上腺素能刺激的心脏反应或药理AMPK激活对收缩功能、钙电流、cAMP含量和腺苷环化酶5(AC5)表达的影响,这是肾上腺素能途径的速率限制步骤。在AMPKα2基因敲除小鼠中,AC5的表达增加。相反,与未受刺激的大鼠心肌细胞相比,5-氨基咪唑-4-羧酰胺核糖体(AICAR)的药理激活AMPK导致AC5表达下降,cAMP含量降低了40%。最后,在实验性压力过载诱导的心功能障碍中,实验表明AMPK激活下调AC5的表达,减弱肾上腺素能。AMPK和-肾上腺素能途径之间可能参与了一种代偿性能量保留机制。因此,本研究在健康和功能失调的心脏中,AMPK激活和β-肾上腺素能通路之间可能存在的相互作用。
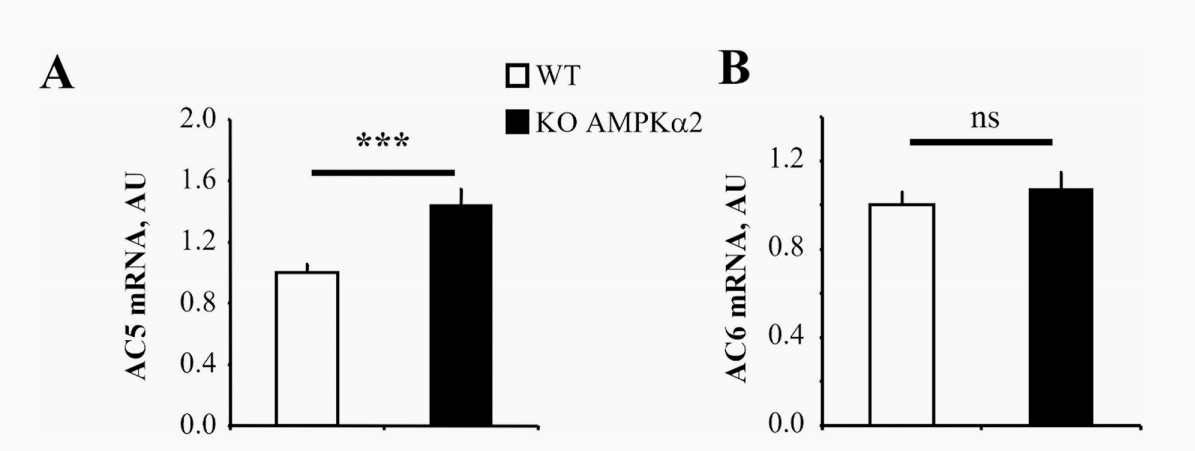
图1:AC5和AC6在WT和AMPKα2-/-小鼠中的表达
参考文献
Modulation of cardiac cAMP signaling by AMPK and its adjustments in pressure overload-induced myocardial dysfunction in rat and mouse
二、TREM2基因敲除细胞
TREM2基因编码一种膜蛋白,该膜蛋白与 TYRO 蛋白酪氨酸激酶结合蛋白形成受体信号转导复合物。编码的蛋白质在免疫反应中发挥作用,并可能通过触发组成型炎性细胞因子的产生参与慢性炎症。该基因的缺陷是导致多囊脂膜性骨发育不良伴硬化性脑白质病的原因。可变剪接导致编码不同亚型的多个转录变体。
今年6月中山大学第一附属医院实验团队发表了一篇研究肝细胞癌经动脉化疗栓塞后,TREM2敲除巨噬细胞在抑制CD8T细胞渗透中的重要作用的研究性论文,对肝细胞癌(HCC)经动脉化疗栓塞(TACE)后的免疫机制进行了探究。
本文研究收集5例未接受治疗的HCC患者和5例接受TACE治疗的患者的肿瘤样本,并对其进行单细胞RNA测序。另外22个配对样本使用免疫荧光染色和流式细胞术进行验证。在TACE治疗后,CD8+ T细胞数量减少,肿瘤相关巨噬细胞数量增加。
TREM2在TACE后的巨噬细胞中高表达。在体外共培养TREM2基因敲除小鼠和野生型小鼠两种模型等试验,即HCC细胞原位注射模型和自发性HCC模型。TREM2的缺失增加了CD8+ T细胞的渗透,从而抑制了两种肝癌模型中的肿瘤生长。更重要的是,TREM2缺乏增强了抗pd - l1阻断的治疗效果。这些发现解释了肝癌TACE后复发的原因,并为肝癌TACE后的免疫治疗提供了新的靶点。
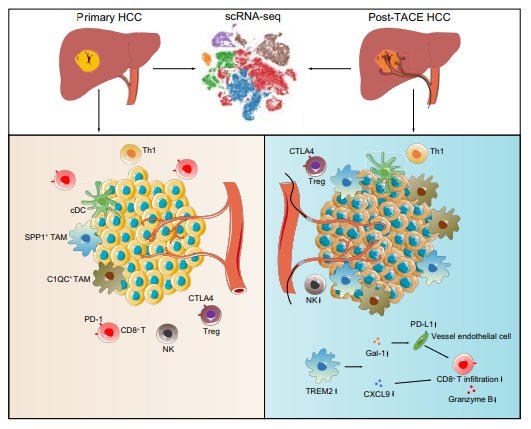
图2:TREM2+ 巨噬细胞抑制肝细胞癌经动脉化疗栓塞术后的 CD8+ T 细胞渗透
参考文献TREM2 macrophages suppress CD8 T-cell infiltration after transarterial chemoembolisation in hepatocellular carcinoma ++
三、P53基因敲除细胞
P53基因编码一种肿瘤抑制蛋白,包含转录激活、DNA结合和寡聚化结构域。编码蛋白响应不同的细胞应激来调节靶基因的表达,从而诱导细胞周期阻滞、细胞凋亡、衰老、DNA修复或代谢变化。该基因的突变与多种人类癌症有关,包括遗传性癌症。该基因的选择性剪接和替代启动子的使用导致多种转录物变体和同工型。
今年7月德克萨斯大学实验团队在Blood Advances杂志上发表的一篇文章,研究证明了p53的缺失增强了克隆源性多发性骨髓瘤细胞的肿瘤起始潜能和耐药性。肿瘤复发和耐药性是限制多发性骨髓瘤(MM)治愈能力的主要因素。p13染色体缺失(del17p)包括TP53基因的缺失,是一种高风险的细胞遗传学异常。
他们发现p53的缺失增加了肿瘤起始MM细胞(TICs)的频率和耐药性。随后的RNA测序(RNA-seq)研究表明,在p53敲除(p53-KO)细胞中,Notch信号通路显著激活,DNA结合抑制剂(ID1/ID2)基因表达上调。他们发现,ID1或HES-1表达的缺失或伽马分泌酶抑制剂(GSI)的处理显著降低了p53-KO的克隆生长,但没有降低p53野生型细胞的克隆生长。在一组MM样本中,GSI处理也降低了del17p样本的克隆生长,但在非del17p样本中没有。因为Notch胞内结构域(NICD)的过表达恢复了GSI治疗的效果。这些机制是否调控p53-KO MM中的Notch1-Hes1信号通路有待进一步研究。他们的研究结果支持了靶向Notch1-Hes1信号通路的潜在可能,包括使用GSIs选择性地减少p53-KO细胞的克隆生长和耐药性。
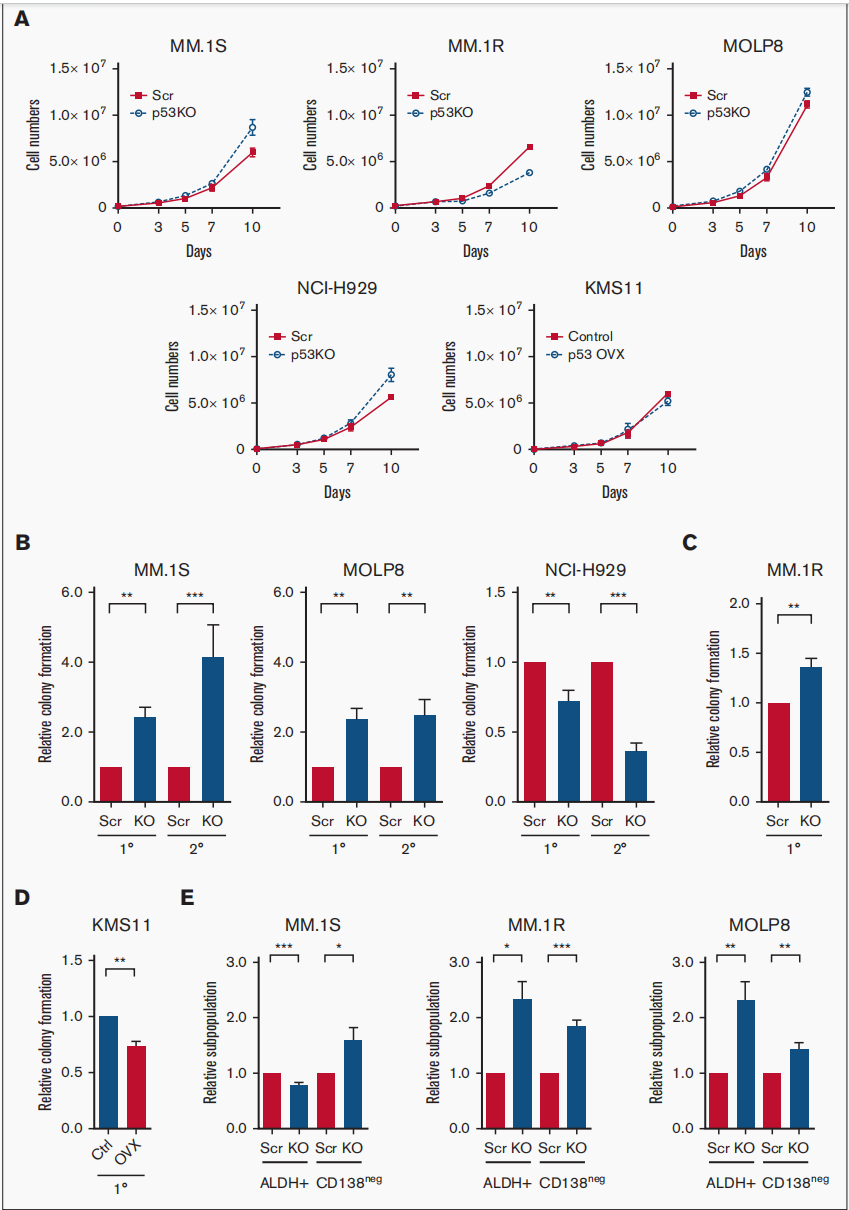
图3:p53的缺失增强了MM细胞的肿瘤起始潜能。
参考文献
Loss of p53 enhances the tumor-initiating potential and drug resistance of clonogenic multiple myeloma cells
★来get大佬同款!艾迪基因提供上述研究的AMPK、TREM2、p53基因敲除细胞现货,快速交付一周送达,低至6800起!
1810225074(微信同号)
market@edgene.cn
最近资讯
【前沿资讯】基因编辑新动向-敲除细胞、先导编辑点突变技术照亮基因研究之路